Abstract
Lennert lymphoma (lymphoepitheloid lymphoma) is an extremely rare variant of peripheral T-cell lymphoma, not otherwise specified. Here we report a case of Lennert lymphoma diagnosed in a 57-year-old woman. She had a three-year history of waxing and waning lymphadenopathy with a rapid increase in size in the past four months before presentation. A needle biopsy and a fine needle aspiration were non-diagnostic due to extensive necrosis. The patient underwent a right neck lymph node excisional biopsy which showed the lymph node architecture was effaced by numerous and sometimes confluent clusters of epithelioid histiocytes and infiltration of small lymphocytes. Extensive necrosis was present. Immunohistochemical stains revealed a mixed population of B- and T-cells with the T-cells showing diminished T-cell markers CD3, CD5, and CD7. Flow cytometric analysis detected a small population (7% of total lymphocytes) of CD4-positive T-lymphocytes with loss of CD3, CD5, and CD7 expressions. PCR-based T-cell receptor gene rearrangement studies showed positive results (clonal peaks) in both gamma and beta genes. Stains for microorganisms were negative. The overall findings indicate Lennert lymphoma. To our knowledge, this is the first reported case of Lennert lymphoma with extensive necrosis. The patient is undergoing chemotherapy. The diagnosis of Lennert lymphoma can be challenging, particularly in cases with extensive necrosis. Our case highlights that adequate sampling is important in the investigation of patients with suspected Lennert lymphoma. A careful pathologic examination with ancillary studies including flow cytometry, immunohistochmistry, and cytogenetic and molecular studies leads to the accurate diagnosis.
Author Contributions
Academic Editor: Rada M. Grubovic, Majka Tereza 17 1000 Skopje Macedonia.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2021 Jiajie G. Lu, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Lennert lymphoma, also known as lymphoepithelioid lymphoma, is a rare variant of peripheral T-cell lymphoma, not otherwise specified (NOS).1 Histologically it is characterized by the presence of two main cell populations: clusters of epithelioid histiocytes and scattered lymphocytes. The lymphocytes are small in size and contain slightly irregular nuclei with coarse chromatin.2 Occasional large Reed-Sternberg-like cells may be present. Necrosis has not been reported as a feature of this lymphoma. Here we report a case of Lennert lymphoma with extensive necrosis. We highlight the diagnostic challenge of approaching such a case and the possible differential diagnoses, and review the clinical, histologic and other diagnostic features of this rare lymphoma.
Case Report
A 57-year-old woman with history of depression presented with diffuse lymphadenopathy of the cervical, axillary, inguinal and left thigh lymph nodes. She first noticed cervical and axillary lymph nodes three years ago that were waxing and waning in course. She decided to seek work-up when her left thigh lymph node began to increase rapidly in size over four months. She denied fevers, chills, or weight loss. Her PET scan showed extensive hypermetabolic lymphadenopathy within the bilateral neck, bilateral axilla, chest, pelvis, left upper thigh, and bilateral inguinal regions; the largest lymph node was located in the left upper thigh, measuring 5.2 cm in greatest dimension, with maximum SUV of 14.4. Her LDH at presentation was 239 U/L (reference range: 98-192 U/L). The left thigh mass underwent 18 gauge core biopsy which showed extensive necrosis surrounded by a rim of lymphoid tissue and was non-diagnostic. A fine needle aspiration of a level V cervical lymph node also yielded non-diagnostic results. Finally, core biopsies from a left groin lymph node using 14 gauge needles and an excisional biopsy of a right neck lymph node were obtained, which provided diagnostic tissues. The two specimens showed similar morphologic and immunohistochemistry features. The H&E stained sections showed the lymph node architecture was effaced by a diffuse lymphohistiocytic infiltrate with extensive geographic necrosis (Figure 1 A and B). There were numerous and sometimes confluent clusters of epithelioid histiocytes with pale reniform nuclei and abundant eosinophilic cytoplasm. Between these histiocytes were many small and a few medium-sized lymphocytes with mature chromatin; some lymphocytes showed slight nuclear irregularities (Figure 1C). Rare eosinophils, rare plasma cells, and occasional scattered immunoblasts were found. Neither high endothelial venules nor Reed- Sternberg-like cells were noted. Gram, Grocott methenamine silver (GMS), periodic acid-Schiff (PAS), and acid-fast (AFB) stains were negative for bacteria, fungal organisms, and acid-fast bacilli, respectively. Flow cytometric analysis performed on the right neck lymph node tissue detected a small population of T-cells (7% of total lymphocytes, 15% of total T-cells) that were positive for CD2 and CD4 but negative for CD3, CD5, CD7, CD8, and CD56. The B cell population was polyclonal. Immunohistochemical stains showed a mixed population of B-cells (CD20 and PAX-5 positive) and T-cells. The T-cells were positive for CD2 (Figure 2A) but had partial loss of CD3 (Figure 2B), CD5 (Figure 2C), and CD7 (Figure 2D) expressions. TdT was negative. CD8-positive T-cells appeared slightly more frequent than CD4-positive T cells. A few scattered lymphocytes were positive for TIA1. Ki67 immunohistochemical stain showed a proliferation rate up to 40%. No Hodgkin Reed-Sternberg cells were identified by CD30, CD20, and PAX-5 stains. In situ hybridization for Epstein–Barr virus-encoded RNA (EBER) was negative. PCR-based T-cell receptor gene rearrangement studies were performed on both left groin and right neck lymph nodes and showed similar clonal peaks in the gamma and beta genes. The histologic and immunohistochemistry features, flow cytometric findings, and molecular results were consistent with Lennert lymphoma. A bone marrow aspiration and biopsy did not reveal evidence of lymphoma in the bone marrow. The patient was determined to have stage III disease with the international prognostic index (IPI) of 2.
Figure 1.Representative photomicrographs of the excisional biopsy of a right neck lymph node. Low power view showing the lymph node architecture was effaced by a diffuse lymphohistiocytic infiltrate (A) with extensive geographic necrosis (B). At high power, the lymphocytes are small to intermediate in size; they have round or oval to slightly irregular nuclei, coarse chromatin, and sometimes small nucleoli (C). (H&E stain; original magnification, x 100 A and B, x 400 C).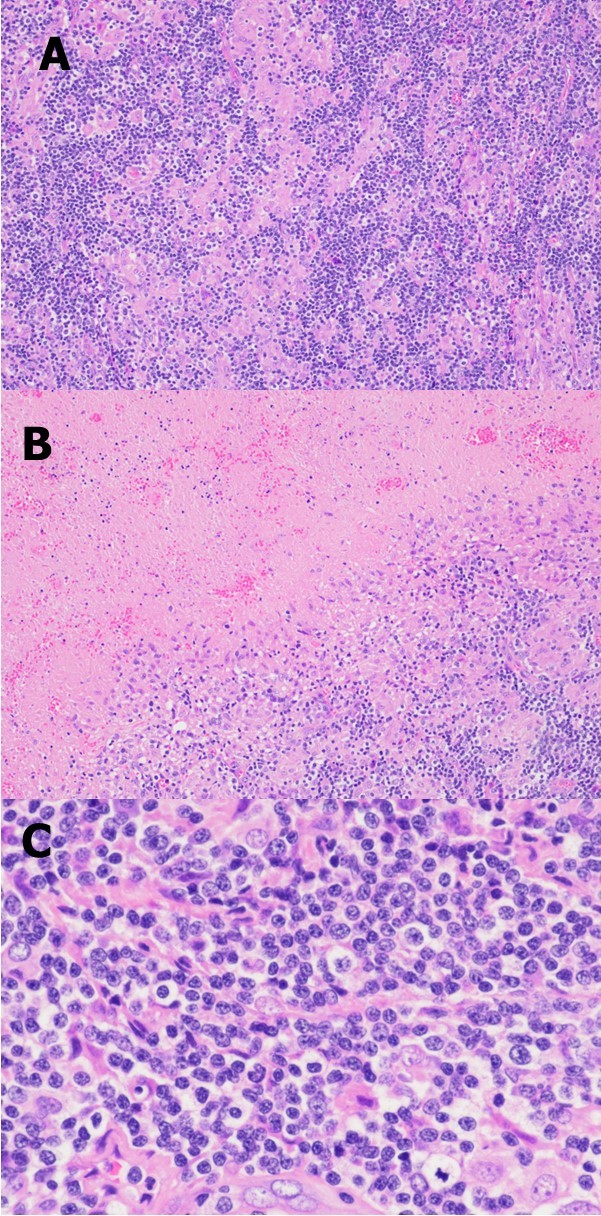
Figure 2.A panel of immunohistochemical stains show the T-lymphocytes are positive for CD2 (A) with partial loss of CD3 (B), CD5 (C), and CD7 (D) expressions. (Immunoperoxidase staining; original magnification, x 200).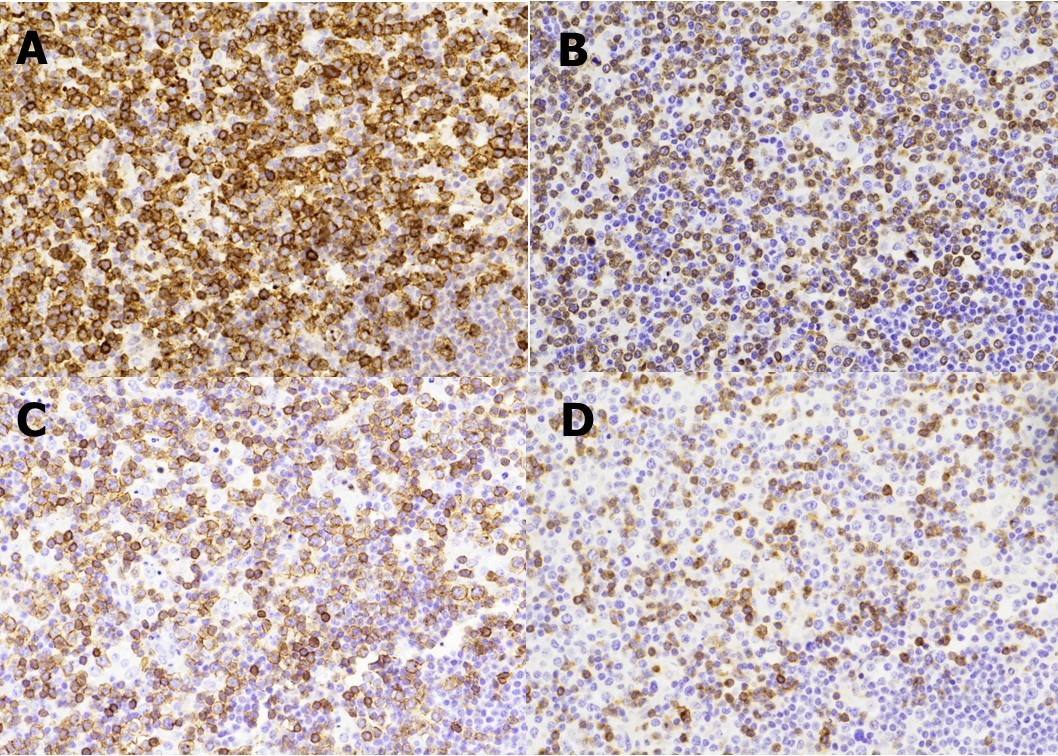
The patient was treated with CHOEP chemotherapy (cyclophosphamide, doxorubicin, vincristine, etoposide, prednisone) with marked reduction in the sizes of her lymph nodes on clinical exam. A PET scan was performed after four cycles of chemotherapy and showed marked interval decrease in size and resolution of activity of multi-station adenopathy. After six cycles of chemotherapy, PET scan was repeated and showed a new 6 mm hypermetabolic soft tissue structure in the right masticator space and new hypermetabolic foci of skin thickening in the left axilla and right gluteal region. A skin shave biopsy showed dermal lymphohistiocytic infiltrate consistent with involvement by Lennert lymphoma. The patient is on clinical trial of duvelisib and is undergoing evaluation for autologous stem cell transplant.
Discussion
Lennert lymphoma was first described by Karl Lennert as a variant of Hodgkin lymphoma.3 Later, Lennert et al. renamed the lesion ‘lymphoepithelioid cellular lymphoma’ and categorized it as a non-Hodgkin lymphoma.4 Further studies showed that this lesion was a T-cell lymphoma,5,6 and the neoplastic nature of the lesion is supported by the fact that T-cell receptor genes are clonally rearranged.7,8 Currently it is regarded as a variant of peripheral T-cell lymphoma, NOS.1
Peripheral T-cell lymphomas as a whole comprise 6% of all non-Hodgkin lymphomas. They arise primarily in middle-aged adults, more often in male patients.1 The clinical presentation is usually advanced-stage disease with B symptoms. Bulky disease is often present. The prognosis is poor with 5-year survival of 20-30%.1 Compared to B-cell lymphomas, T-cell lymphomas are difficult to treat, and cytotoxic therapy often does not work or does not have a durable response.9 Often bone marrow transplant is the best treatment.9
Lennert lymphoma is characterized by effacement of lymph node architecture with small lymphocytes showing mild cytologic atypia and prominent, confluent clusters of epithelioid histiocytes.2 Scattered larger lymphoid cells may be present, as are inflammatory cells. There can also be scattered Reed-Sternberg-like B-cells (usually positive for EBV), which were not found in our case. Extensive necrosis, which was seen in the present case, has not been reported.10-12 The extensive necrosis could be caused by an accelerated growth of the lymphoma or could be related to bulky disease. Although it has been reported that spontaneous extensive necrosis in lymphoma is associated with significantly higher stages, greater IPI, and higher serum LDH levels,13,14 the prognostic significance of extensive necrosis in lymphoma is unclear. As a T-cell lymphoma, the neoplastic cells in Lennert lymphoma may show loss of T-cell markers with loss of CD5 and/or CD7 most common.
The cell of origin of Lennert lymphoma is still controversial. Some studies showed the proliferating cells were mainly CD4 positive, indicating a helper T-cell origin.8,15 Other studies concluded Lennert lymphomas are often derived from CD8-positive cytotoxic T-cells. It is likely that the neoplastic cells in some Lennert lymphoma cases are of CD4-positive helper T-cell origin while in other cases are derived from CD8-positive cytotoxic T-cells. In our case the lymphoma cells were positive for CD4 by flow cytometry.
It is difficult to make a diagnosis of Lennert lymphoma as the neoplastic lymphocytes are scattered in a background of many benign/reactive small lymphocytes and show minimal cytologic atypia. In addition, histiocytic proliferation is non-specific and can be seen in many reactive and neoplastic conditions. The presence of extensive necrosis in our case made the diagnosis even more challenging because proliferation of histiocytes with extensive necrosis is more commonly seen in infections, inflammations, and other reactive conditions. Clinicians and pathologists should be aware of this entity and the fact that extensive necrosis does not exclude this entity. The key diagnostic clues for this case are the epithelioid histiocyte proliferation, loss of T-cell markers including CD3, CD5, and CD7, and evidence of T-cell clonality by T-cell receptor gene rearrangement studies. The differential diagnoses might include angioimmunoblastic T-cell lymphoma, classic Hodgkin lymphoma, and T-cell/histiocyte-rich large B-cell lymphoma. In this case we did not see the high endothelial venules characteristic of angioimmunoblastic T-cell lymphoma. Although CD30 stained scattered immunoblasts, diagnostic Hodgkin Reed-Sternberg cells were not identified by either morphology or immunohistochemistry. T-cell/histiocyte-rich large B-cell lymphoma was ruled out given lack of CD20-positive large B-cells. Moreover, because of the extensive necrosis, other conditions such as Kikuchi-Fujimoto disease and infections or inflammations should have also been considered as differential diagnoses. In our patient, mycobacterial and fungal cultures were negative, and no microorganisms were detected by Gram, AFB, GMS, and PAS stains.
Very few Lennert lymphoma cases have been studied by cytogenetic analysis. Although aberrations of chromosome No. 3 (trisomy 3, break in q22, or dup q22 → q24) were reported in six of seven Lennert lymphoma cases,16 this finding is not specific and can be seen in other types of lymphomas. However, cytogenetic abnormalities help establish clonality and support diagnoses of malignancy.
In conclusion, as far as we know, this is the first report on Lennert lymphoma with extensive necrosis.
Author Contributions Statement
JL and CY identified the case. JL performed the literature search and wrote the first draft of the article. PP wrote the clinical history. JC prepared the figures/figure legends and helped shape the manuscript. CY provided critical feedbacks. XQ supervised this work, performed additional literature search, and created the final version of the article.
References
- 1.Swerdlow S H, Campo E, Harris N L, Jaffe E S. (2017) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Stylus Publishing, LLC. , Herndon, VA: 403-407.
- 2.Suchi T, Lennert K, Tu L Y, Kikuchi M. (1987) Histopathology and immunohistochemistry of peripheral T cell lymhomas: a proposal for their classification. , J ClinPathol ; 40, 995-1015.
- 3.Lennert K, Mestdagh J. (1968) Hodgkin’s disease with constantly high content of epitheloid cells. Virchows Arch APatholPatholAnat;. 344, 1-20.
- 4.Lennert K, Mohri N, Stein H, Kaiserling E. (1975) The histopathology of malignant lymphoma. , Br J Haematol; 31, 193-203.
- 5.Palutke M, Varadachari C, Weise R W, Husain M. (1978) Lennert’s lymphoma, a T-cell neoplasm. , Am J ClinPathol; 69, 643-45.
- 6.Han T, Barcos M, Yoon J M, Rakowski I. (1980) Malignant lymphoma with a high content of epithelioid histiocytes: report of a T-cell variant of so-called Lennert lymphomas and review of the literature. , Med PediatrOncol; 8, 227.
- 7.O’Connor N T, Feller A C, Wainscoat J S, Gatter K C. (1986) T-cell origin of Lennert’s lymphoma. , Br J Haematol; 64, 521-8.
- 8.Feller A C, Griesser G H, Mak T W, Lennert K. (1986) Lymphoepithelioid lymphoma (Lennert’s lymphoma) is a monoclonal proliferation of helper inducer T-cells. , Blood; 68, 663-7.
- 9.Laribi K, Alani M, Truong C, A Baugier de Materre. (2018) Recent advances in the treatment of peripheral T-cell lymphoma. , The Oncologist; 23, 1-15.
- 10.Parimal S, Pai R, Manipadam M T, Nair S H. (2013) Lennert’s Lymphoma: Clinicopathological profile of five cases. , Indian J PatholMicrobiol; 45, 248-51.
- 11.Patsouris E, Engel hard M, Zwingers T, Lnnert K. (1993) Lymphoepithelioid cell lymphoma (Lennert’s lymphoma): clinical features derived from analysis of 108 cases. , Br J Haematol; 84, 346-48.
- 12.Kurita D, Miyoshi H, Yoshida N, Sasaki Y. (2016) A clinicopathologic study of Lennert Lymphoma and possible prognostic factors; the importance of follicular helper T-cell markers and the association with angioimmunoblastic T-cell lmpoma. , Am J SurgPathol; 40, 1249-60.
- 13.Abd-Elrahman I, Rapoport V, Neuman T, Appelbaum L. (2010) Clinical and Molecular Significance of Tumor Necrosis. In Newly Diagnosed Patients with Hodgkin's Lymphoma and Diffuse Large B Cell Lymphoma. Blood; DOI: 10.1182/blood.V116.21.1995.1995 116(21).
- 14.Saito A, Takashima S, Takayama F, Kawakami S. (2001) Spontaneous extensive necrosis in non-Hodgkin lymphoma: prevalence and clinical significance. , J Comput Assist Tomogr; 25(3), 482-6.